
Validate bulk RNA-seq inputs | Codex use cases
Codex use cases


Codex use case
Validate bulk RNA-seq inputs
Validate bulk RNA-seq inputs before differential expression.
Difficulty Intermediate
Time horizon Long-running
Use Codex with the NGS Analysis plugin to validate sample sheets, FASTQs, and references, then return MultiQC, Salmon matrices, provenance, and a short QC interpretation before differential expression.
Best for
- Bioinformatics teams validating bulk RNA-seq inputs before differential expression.
- Researchers who want transcript and gene-level quantification plus QC in one thread.
- Teams that need mapping-rate, duplication, library-type, and resource-readiness review.
Contents
Copy page Export as PDF
Use Codex with the NGS Analysis plugin to validate sample sheets, FASTQs, and references, then return MultiQC, Salmon matrices, provenance, and a short QC interpretation before differential expression.
Intermediate
Long-running
Related links
Request access to GPT-Rosalind
Best for
- Bioinformatics teams validating bulk RNA-seq inputs before differential expression.
- Researchers who want transcript and gene-level quantification plus QC in one thread.
- Teams that need mapping-rate, duplication, library-type, and resource-readiness review.
Skills & Plugins
-
Validate sequencing inputs, run bulk RNA-seq counts and QC, and return auditable artifacts.
| Skill | Why use it |
|---|---|
| NGS Analysis | Validate sequencing inputs, run bulk RNA-seq counts and QC, and return auditable artifacts. |
Starter prompt
Use the NGS Analysis plugin. Run bulk RNA-seq FASTQ-to-count QC on the provided sample sheet, FASTQ root, transcriptome FASTA, genome FASTA, and GTF. Return:
- run_manifest.json
- MultiQC plus browser-safe review links
- Salmon transcript- and gene-level matrices
- validation and resource-readiness artifacts
- a short QC interpretation that calls out mapping rate, duplication, library-type agreement, outlier samples, and anything that would block downstream differential expression
Open in the Codex app
Use the NGS Analysis plugin. Run bulk RNA-seq FASTQ-to-count QC on the provided sample sheet, FASTQ root, transcriptome FASTA, genome FASTA, and GTF. Return:
- run_manifest.json
- MultiQC plus browser-safe review links
- Salmon transcript- and gene-level matrices
- validation and resource-readiness artifacts
- a short QC interpretation that calls out mapping rate, duplication, library-type agreement, outlier samples, and anything that would block downstream differential expression
Leverage skills
The NGS Analysis plugin includes:
ngs-analysis-routerngs-bulk-rnaseq-counts-qcngs-runtime-env
When you use the plugin, Codex can use all these packaged skills.
Step-by-step guide
- Point Codex to a directory with the sample sheet, FASTQs, transcriptome FASTA, genome FASTA, and GTF, or provide exact file references.
- Run the starter prompt so Codex can validate strandedness, reference consistency, and tool readiness before execution.
- Open the generated MultiQC and matrix artifacts in Codex to review mapping rate, duplication, library-type agreement, and resource readiness.
- Continue in the same thread to fix blockers, rerun with updated metadata, or hand the resulting gene-level matrices into downstream differential expression.
Results
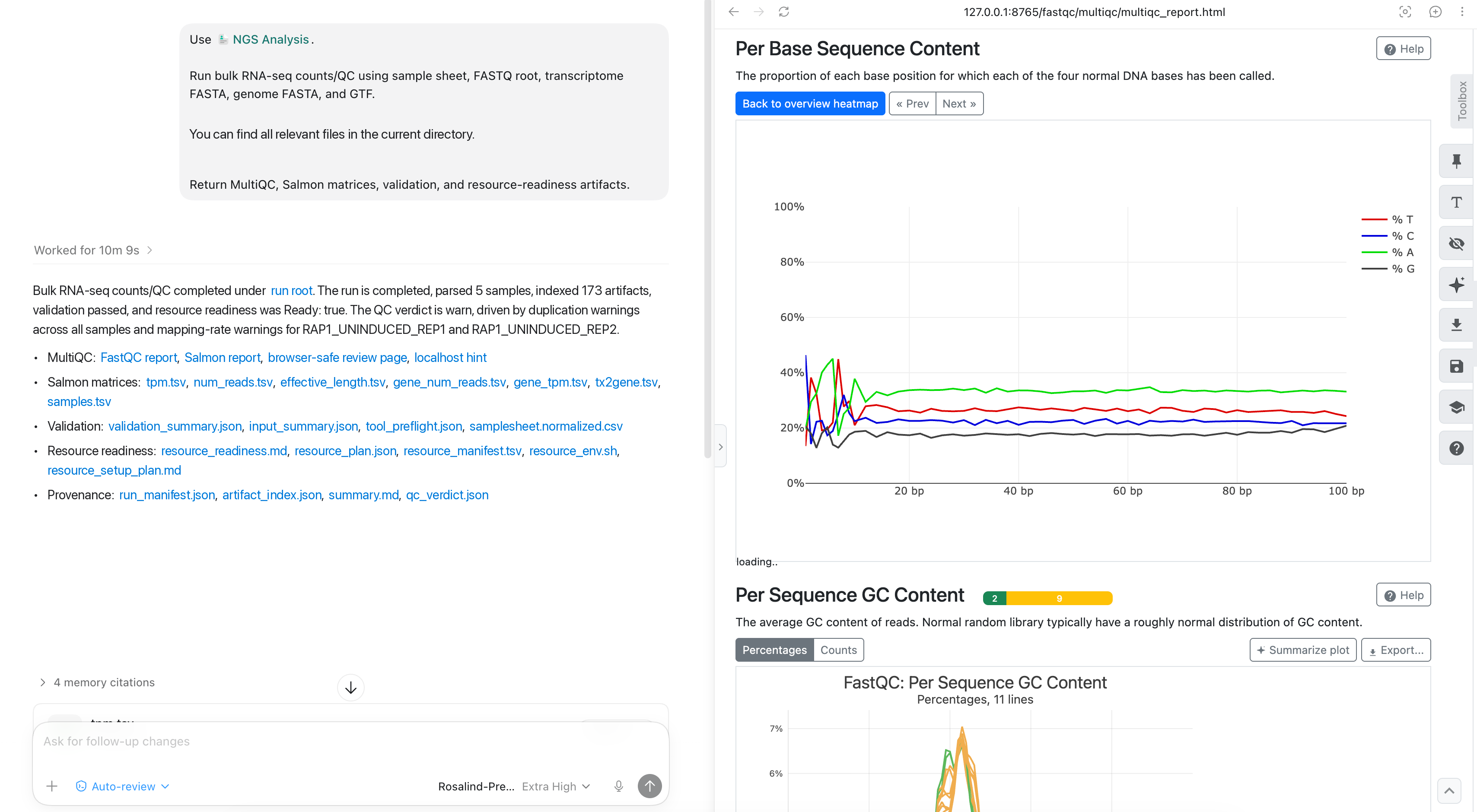
The run returns a QC-reviewed counts bundle rather than a bare quantification output. Start with the MultiQC report to identify warnings that could affect downstream interpretation. In this example, Codex surfaces FastQC sequence-content warnings alongside the run summary so the team can decide whether the observed pattern is expected for the library preparation.
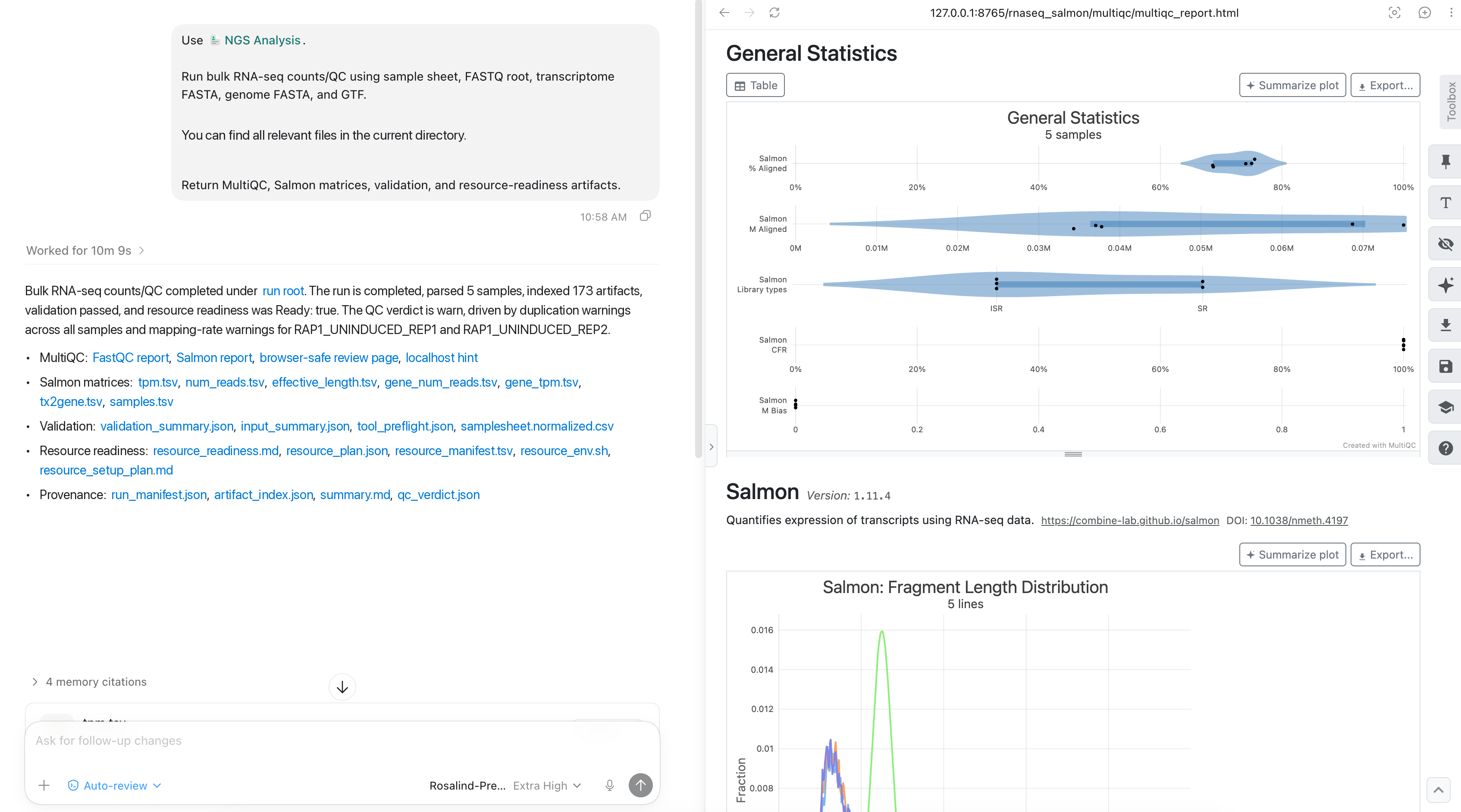
Next, review the Salmon statistics in the same report. Mapping rates, library-type assignments, and duplication signals provide a compact readiness check before differential expression.
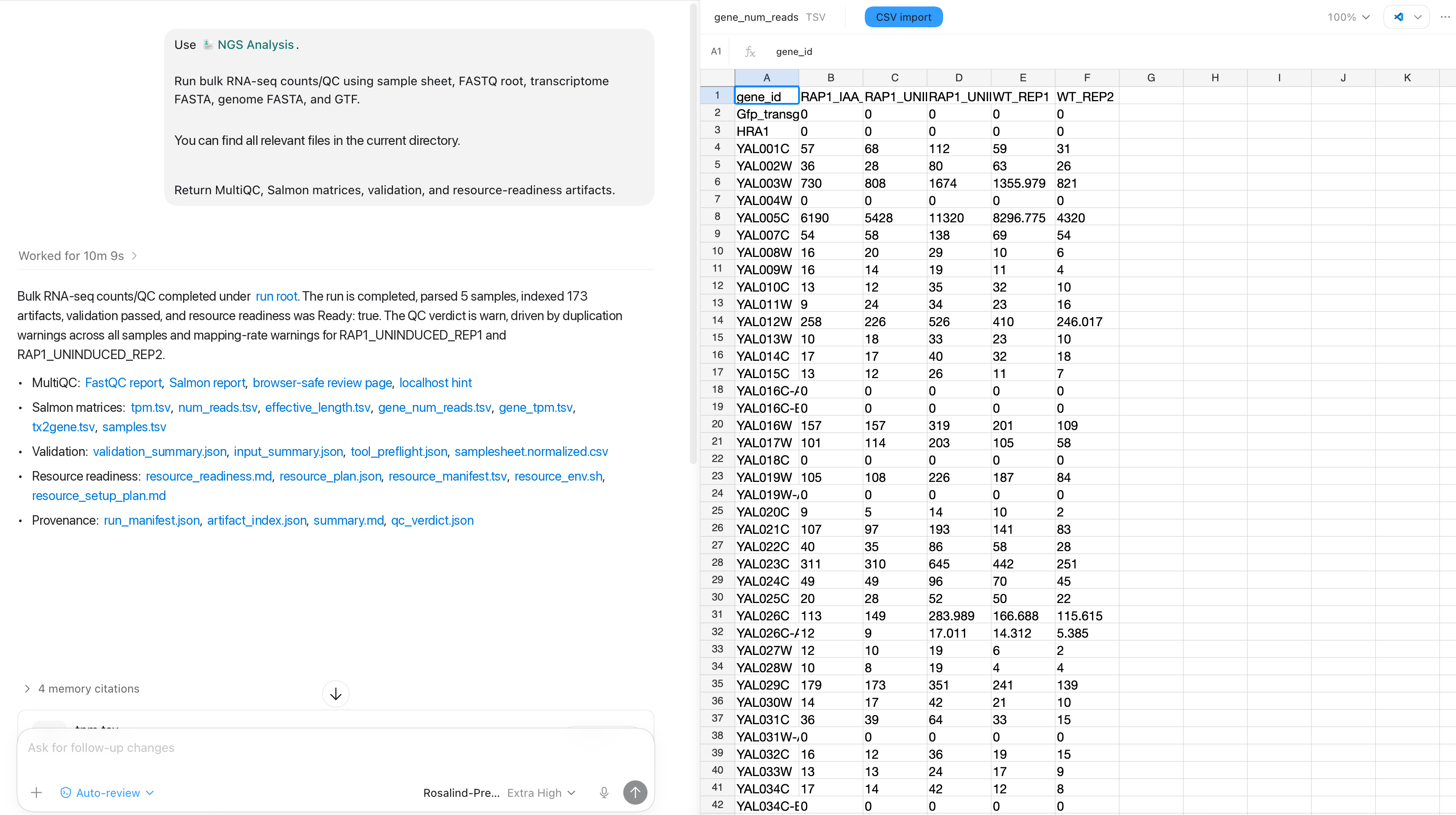
The resulting gene-level count matrix is saved as a reusable artifact. Open it in Codex to confirm the expected samples and features are present, then keep it with the run provenance for downstream analysis.
Related use cases
[
Annotate scRNA-seq data
Use Codex with the NGS Analysis plugin to turn a 10x-style matrix bundle into QC-filtered...
Sciences Data](https://developers.openai.com/codex/use-cases/scrna-seq-post-count-qc)[
Discover protein folding architectures
Use Codex with Goal Mode to research and implement novel architectural modifications to...
Sciences Engineering](https://developers.openai.com/codex/use-cases/discover-protein-folding-architectures)[
Prioritize drug targets
Use Codex with the Life Science Research plugin to normalize entities, retrieve genetics...
Sciences Data](https://developers.openai.com/codex/use-cases/target-prioritization)